What are Hemorrhagic Disorders?
Introduction
The primary function of hemostasis is to stop bleeding at the site of an injury and at the same time to maintain the normal fluid state of blood in the circulatory system. The bleeding is stopped by the formation of a hemostatic plug or network with help of platelets (thrombocytes) and various clotting factors (proteins)1. Formation of this network is completed through the intrinsic and extrinsic pathways of coagulation, both of which trigger the final, common steps (common pathway) involved in the conversion of fibrinogen into fibrin. There should be a delicate balance between hemorrhage and thrombosis for normal hemostasis. Any imbalance can lead to hemorrhagic disorders (bleeding disorder) or thrombotic disorder (clotting disorder)(2).
Bleeding can occur due to many reasons, they being: very few or abnormal platelets, abnormal or low amounts of clotting proteins, or abnormal blood vessels. Normally, in a bleeding disorder, the first two stages of hemostasis function normally – the blood vessels constrict, an immature platelet plug is formed, and bleeding may stop or slow down. But in an abnormal situation, since one of the clotting factors is decreased or absent, a mature fibrin clot is not formed; instead, a soft, ineffective clot is formed. The soft platelet plug breaks down, the bleeding resumes, and the process begins again, leading to repeated cycles of bleeding and partial clotting. Following are most common bleeding disorders(2):
-
Hemophilia A or B: Factor VIII and Factor IX are absent in this disorder.
-
Von Willebrand Disease (VWD): Von Willebrand Factor is absent in this disorder.
-
Rare Bleeding disorders: One of the other 8 clotting factors is decreased or absent.
Hemophilia A (factor VIII deficiency) and hemophilia B (factor IX deficiency) are X chromosome linked recessive congenital bleeding disorders, hence primarily occurs in males (Figure 1). Hemophilia A prevalence is 1:10,000 live male births, and hemophilia B is 1:30,000. In rare cases, factor XI deficiency was classified as hemophilia C and acquired hemophilia resulting from the development of autoantibodies against factor VIII or rarely IX, in a patient with no previous bleeding history. It affects males as well as females.
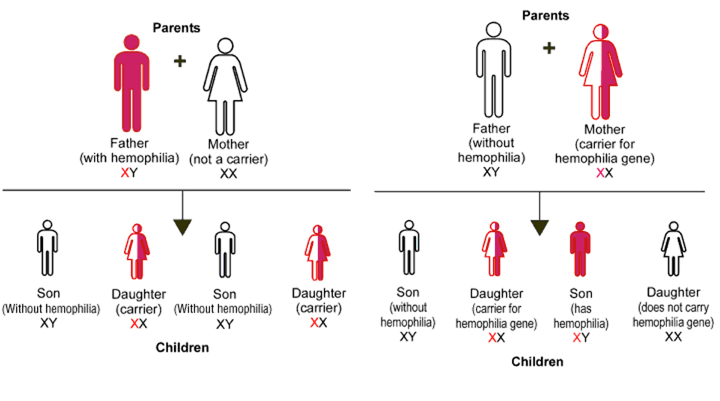 Figure 1: Inheritance of Hemophilia
Figure 1: Inheritance of Hemophilia
(Source: http://www.daviddarling.info/encyclopedia/H/hemophilia.html)
A male with hemophilia shall have personal history of spontaneous major bleeding episodes (mostly in joints, skeletal muscles, gastrointestinal tract, and occasionally in the central nervous system or in other organs). Repeated episodes of joint bleeding may result in chronic disability or even fatalities if not treated in time. In infants bleeding at circumcision, on tooth eruption, or when first standing (in knee joints) should raise the suspicion of hemophilia. The severity of bleeding is similar for the same levels of factor deficiency in hemophilia A and B3.
Laboratory assessment of Hemophilia
-
Screening tests: Platelet counts, PT (Prothrombin Time), and PTT (partial thromboplastin time) are most commonly recommended as a screening test for patients with a bleeding medical condition. In hemophiliacs, platelet count and PT are normal and PTT is prolonged.
(ECL Note: ECL series has primary test menu which starts with Erba PT LS (EHL00023) (For PT assay) and Erba Actime (EHL00003) (For PTT assay))
-
Confirmatory tests: In severe hemophiliacs Factor VIII or IX levels are below 1%, moderate hemophiliac have factor levels between 1% and 5%, and the mild cases >5%. These percentages can be obtained by quantitative estimation of Factor VIII and IX.
(ECL Note: ECL series has Erba Factor VIII deficient plasma (EHL00033) (For Factor VIII estimation) and Erba Factor IX deficient plasma (EHL00033) (For Factor IX estimation). These assays can be calibrated by using Erba Standard Plasma (EHL00012). Whereas, Erba Control N Plus (EHL00016) and Erba Control P Plus (EHL00017) can be used for quality control of these assays.)(3)
Von Willebrand Disease (VWD) is a genetic disorder caused by abnormal Von Willebrand Factor (VWF); causing excessive mucocutaneous bleeding. VWF binds to factor VIII, and platelets in blood vessel walls, which helps in platelet plug formation during the clotting process. VWD is the most common bleeding disorder, seen up to 1% of the Caucasian population. The related gene is situated on chromosome 12 and this disorder occurs equally in men and women.
The common symptoms for VWD are:
-
Bleeding nose
-
Easy bruising
-
Excessive bleeding during and after invasive procedures, such as tooth extractions surgery
-
Heavy menstrual bleeding in women
-
Hemorrhaging after childbirth.
There are three main types of VWD based on qualitative or quantitative defects in VWF. A fourth type, is acquired VWD and is not hereditary.
-
Type 1 VWD is found in 60%-80% of patients. Symptoms are usually mild.
-
Type 2 VWD is found in 15%-30% of patients. Symptoms are mild to moderate.
-
Type 3 VWD is found in 5%-10% of patients. Symptoms are typically severe, and include spontaneous bleeding episodes, often into their joints and muscles.
-
Acquired VWD. This type of VWD in adults results after a diagnosis of an autoimmune disease, such as lupus, or from heart disease or some types of cancer. It can also occur after taking certain medications.
Laboratory assessment of VWD:
-
Initial assessment can begin with a platelet function test, as the clinical manifestations of VWD and platelet defects are similar.
-
First-tier tests:
-
vWF Ag (vWF:Ag)
-
Factor VIII coagulant.
(ECL Note: Transasia’s ECL series has Erba Factor VIII deficient plasma (EHL00033) (For Factor VIII estimation). These assays can be calibrated by using Erba Standard Plasma (EHL00012). Whereas, Erba Control N Plus (EHL00016) and Erba Control P Plus (EHL00017) can be used for quality control of these assays.)3
-
Ristocetin cofactor (vWF:RCo) assay measures vWF activity. A ratio vWF:RCo/vWF:Ag <0.7 is indicative of a qualitative vWF defect.
-
VWF activity: A new latex immunoassay for the quantitation of vWF
-
The collagen-binding assay is another functional assay used by some laboratories.
- Second-tier tests:
- VWF multimers is a useful assay to determine various subtypes of the disease.
- Ristocetin-induced platelet aggregation (RIPA). In this assay, the patient’s platelets and plasma are used as a source of vWF.
- Genetic tests.
Rare inherited bleeding disorders (RBDs) includes deficiencies of coagulation factors fibrinogen, factor II, FV, combined FV and FVIII, FVII, FX, FXI, FXIII, and congenital deficiency of vitamin K-dependent factors (VKCFDs). All these disorders are transmitted as autosomal recessive conditions; except FXI and dysfibrinogenemia may be autosomal dominant. The incidence of RBDs are reported with homozygous or a double heterozygous incidence varying from 1 in 500 000 for FVII deficiency to 1 in 2 to 3 million for prothrombin and FXIII deficiencies4.
Initial screen for RBD shall be done by coagulation screening tests including the Thrombin Time (TT), APTT and PT.
Table 1 - General feature of deficiency with respect to coagulation testings
|
|
Laboratory Diagnosis
|
|
Deficiency
|
TT
|
APTT
|
PT
|
|
Fibrinogen
|
↑↑
|
↑↑
|
↑↑
|
|
Prothrombin
|
Normal
|
↑
|
↑
|
|
Factor V
|
Normal
|
↑
|
↑
|
|
Combined Factor V and VIII
|
Normal
|
↑
|
↑
|
|
Factor VII
|
Normal
|
Nomal
|
↑
|
|
Factor X
|
Normal
|
↑
|
↑
|
|
Factor XI
|
Normal
|
↑
|
Normal
|
|
Factor XIII
|
Normal
|
Normal
|
Normal (Specific assay required)
|
|
VKDCF
|
Normal
|
↑
|
↑↑
|
A prolonged APTT with a normal PT suggests FXI deficiency after exclusion of FVIII, FIX, and FXII deficiencies. The reverse pattern is typical of FVII deficiency, whereas the prolongation of both tests directs further analysis toward deficiencies of combined FV and FVIII, FX, FV, prothrombin, or fibrinogen. All coagulation tests depending on the formation of fibrin as the end point are necessary to evaluate fibrinogen deficiency; hence, beside the PT and APTT, the TT is performed (Table 1).
(ECL Note: ECL series has
-
Erba Factor V deficient plasma (EHL00030) (For Factor V estimation)
-
Erba Factor VIII deficient plasma (EHL00033) (For Factor VIII estimation)
-
Erba Factor VII deficient plasma (EHL00031) (For Factor VII estimation)
-
Erba Factor X deficient plasma (EHL00032) (For Factor X estimation)
-
Erba Factor XI deficient plasma (EHL00035) (For Factor XI estimation)
-
These assays can be calibrated by using Erba Standard Plasma (EHL00012). Whereas, Erba Control N Plus (EHL00016) and Erba Control P Plus (EHL00017) can be used for quality control of these assays.)(3)
References
-
Andrew J. Gale; Current Understanding of Hemostasis; Toxicol. Pathol. 2011 ; 39(1): 273–280
-
Regina B. Butler; Introduction to Bleeding Disorders; Nursing Working Group – Nurses’ Guide to Bleeding Disorders; 2012
-
Mary Williamson. L. Micheal Snyder; Interpretation of Diagnostic Tests, Tenth Edition. Wolters Kluwer.
-
Roberta Palla; Rare bleeding disorders: diagnosis and treatment; Blood, 26 March 2015 x Volume 125, Number 13